
Novel bioluminescent reporters
Finding, testing and generating optimised bioluminescent reporter vectors for use in various organisms.
The Idea
Bioluminescent reporters are a common tool in molecular biology. Luciferases from various sources (firefly, click beetle, dinoflagellate, sea pansy, copepod and bacterial) have been cloned and exploited as reporters, due to their fast time dynamics and their detectable and quantifiable outputs. Each luciferase has particular enzymatic properties and substrate requirements, leading to limitations on how assays can be performed. While all eukaryotic luciferases require the addition of an exogenous substrate (eg. luciferin, calcium ions) to produce light, bacterial bioluminescence is generated by a single lux operon which has the benefit of autonomous luminescence [Engebrecht et. al, Cell 1983].
Yet, compared to fluorescent protein reporters, little engineering has been applied to improve and optimize the lux operon on a protein level. Our idea is to sequence the genomes of various wild free-living marine bioluminescent bacterial species, in order to discover novel genes and gene variants responsible for bioluminescence and use these as novel luminescent reporters.
Seminal research has demonstrated important aspects about the structure of the lux operon in bioluminescent bacteria and has highlighted fundamental regulatory elements for light production
[Bassler et al., Mol Microb. 1993; Fuqua et al., J Bacteriol. 1994]. Recently, the 2010 Cambridge iGEM team provided an excellent demonstration of the use of synthetic biology practices to implement lux operon derived bacterial bioluminescence in E. coli. The team expressed a codonoptimized lux operon cassette driven by the arabinose inducible pBAD promoter, free of the endogenous quorum sensing regulation, and they were able to achieve high levels of tunable bioluminescence.
Although research has focused on symbiotic bioluminescent bacteria, many freeliving marine bacteria exhibit bioluminescence to a highly variable degree. Whereas some strains can be seen only after several minutes in a dark room, bright strains are distinguishable by a blue glow even in low light levels. Bacterial bioluminescence has been studied since the late XIX century [Beijerinck, Arch Neerl des Sci Exact et Nat 1889], however, there have been few attempts to systematically quantify the luminosity of freeliving environmental bioluminescent bacterial strains and search for the underlying genetic differences that might account for this variance of luminosity. Our plan is to systematically study a collection of such bacteria that have been gathered and isolated from marine environments around the world, in order to gain insight into their light producing mechanisms and generate reporters.
After sequencing the genomes and finding the sequences of bioluminescent genes, we plan to test them in a standardised E. coli context, in order to gain insight into the structurefunction relationships. On the basis of this data, we will then generate optimised bioluminescent reporter vectors for use in various organisms.
The Team


Bernardo Pollak
Graduate Student, Department of Plant Sciences
Anton Kan
Graduate Student, Department of Plant Sciences
Project Outputs

Project Report
Summary of the project's achievements and future plans

Project Proposal
Original proposal and application
Download PDF >>

Project Output
Follow-up project
Finding, testing and generating optimised bioluminescent reporters.

Figure 1. Environmental bioluminescent bacteria isolation. A, Plate after O/N incubation at RT plated with 1mL of shore seawater. B, Propagation plate with streaked colonies for isolation of bioluminescent bacteria. C, Gram strain micrograph of an isolated strain (63x). D, Multiple isolated bioluminescent bacteria streaked on a single plate for cross-comparative visualization of bioluminescence levels.
Outcomes, Outputs and Progress
Shore seawater was sampled from the Pacific, Atlantic, Caribbean oceans and the Mediterranean sea and 1mL of seawater was plated on LSW-70 agar plates. Colonies were then screened for luminescence and luminescent colonies were streaked out into new plates progressively until single luminescent colonies were distinguishable (Figure 1). Colonies were propagated into liquid media and stored at -80ºC in in 20% glycerol stocks. Species were identified by 16S sequencing (Figure 2).

Figure 2. 16S phylogeny for isolated strains. A phylogenetic tree was produced using the 16S sequences of the strains by using the Geneious tree builder on a Global-alignment with free-end gaps alignment type with a 65% similarity matrix by the neighbour-joining method and the Tamura-Nei genetic distance model.
For each strain, a single colony was inoculated into 10mLs of LSW-70 media and cultures were grown overnight at room temperature. Genomic DNA was extracted using the Life Technologies Genomic Purelink Genomic DNA extraction kit according to manufacturer’s instructions and DNA concentration was measured using the QuBit dsDNA HS Assay kit. Samples were diluted to 2.5ng/uL and sent for library preparation and Illumina sequencing. Library preparation was carried out using the Nextera XT DNA Library Preparation Kit and paired-end sequencing was performed on an Illumina Miseq platform using the 500 cycles v.2 program (2x250bp). Reads were screened and trimmed of adapter contamination using the bbduk command-line program from the bbtools open-source software package with the following parameters:
$bbduk.sh -Xmx1g in1=<reads_1.fq> in2=<reads_2.fq> out1=<filtered1.fq> out2=<filtered2.fq> ref=<adapters.fa> ktrim=r k=23 mink=11 hdist=1 tpe tbo
Filtered reads were then assembled using the SPAdes 3.5.0 genome assembler (Bankevich et al., 2012) with parameters for long-reads:
$python spades.py -o new_assembly -t 16 --pe1-1 <trimmed1.fq> --pe1-2 <trimmed2.fq> --careful
Assembled contigs were then blasted to a blastn database containing curated lux operons from a range of bioluminescent bacteria and hits were screened for ORFs on the Biomatters Geneious 8.1.2 software by the annotate from database function against the curated lux operon set. Sequences were then aligned by pairwise alignment using the Geneious alignment algorithm for global alignment with free-end gaps using a 65% similarity matrix.
Whole lux operons and luxAB genes were then cloned into a pSB3K3 vector by Gibson assembly, driven by a constitutive promoter J23101 (Biobricks registry). We also generated a restructured operon construct, in which we have split the luciferase genes (luxAB) from the substrate synthesis genes (luxCDEG or luxCDEFG) into separate operons on the pSB3K3 vector, driven by the R0011 and J23101 promoters respectively (Biobricks registry).
Screening of positive clones on agar plates and measuring of luminescence was performed on a refurbished gel documentation box mounted with the Photometrics Evolve 512 EMCCD camera using a Nikon 15-80mm camera lens controlled with the Micro-Manager open-source microscopy software. Imaging settings for luminescence measurements were 2000 ms of capture time with 500 EMCCD gain, and images were captured with the box door half open and completely closed to obtain both bright images of the plate and colonies positions and luminescence from lux operon/luciferase expressing colonies (Figure 3).
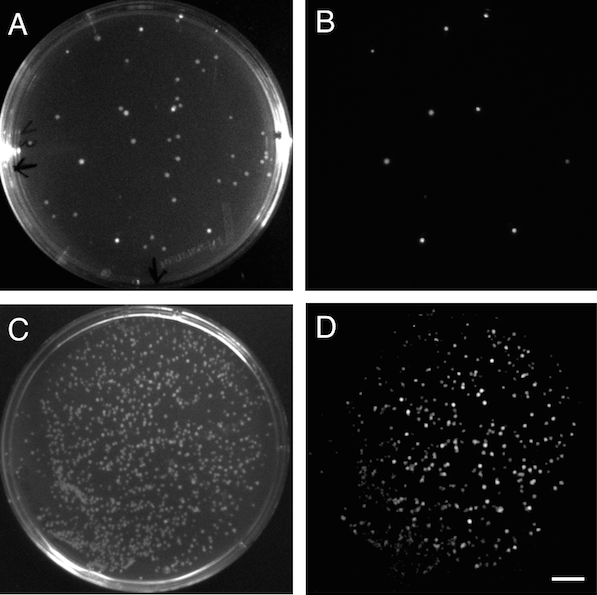
Figure 3. Bioluminescence in E. coli. A, bright image of a plate with E. coli transformants after cloning of a lux operon to the pSB3K3 vector. B, Luminescent positive clones expressing the cloned lux operon. C, bright image of a E. coli transformants after a round of evolution of the lux operon. D, luminescent colonies showing variable brightness due to different evolved lux operons being expressed. Scale bar (in D) measures 1 cm.
Luminescence yield from the bacteria was quantified more accurately in a Clariostar plate reader (BMG Labtech), in which cells were grown overnight and their luminescence and absorbance measured every 10 minutes. The environmental bacteria were quantified in the plate reader in LSW-70 media at 25°C. This found a 2000-fold variation in maximal luminescence yield per cell density between the brightest and weakest strains. The operons and luciferases were also measured in E. coli on the pSB3K3 vector, grown at 30°C in M9 minimal media supplemented with 0.2% casamino acids and 0.4% glucose, with the luciferase-only constructs being supplemented with 0.1% decanal as a substrate.
The bioinformatics analysis also revealed three main types of operons from our sequenced genomes, with at least 58% sequence homology, as determined by the Clustal Omega algorithm (Sievers et al., 2011). This provides a rich pool of sequence variation whilst maintaining enough homology for a recombination based directed evolution approach to create a range of novel luminescent constructs. The evolution was performed by the DNA-shuffling method, in which DNA fragments from each lux operon were digested by DNAseI, and reassembled randomly by a primerless PCR, followed by a primer based PCR to obtain fragments to insert into the plasmid backbone. Bands showing the expected size were gel purified and cloned into the pSB3K3 vector through Gibson assembly and transformed into E. coli (Figure 3.C, 3.D). The colonies showed a high amount of variation in luminescence, and the brightest colonies were then selected. Sanger sequencing verified that the evolved constructs contain segments of several operons. We are currently performing more rounds of evolution to select for even brighter constructs.
Conclusions
Our project has successfully isolated, sequenced and cloned the lux operons from 19 diverse marine environmental bacterial species. The bioinformatics results have shown that we have three main clusters of lux operons, with significant variation within each cluster. These variations also broadly correlate with a wide range of luminescence yield from the environmental strains. We cloned all the operons into an E. coli context, and performed quantification in a better defined DNA context, which found luminescence from all of our constructs. The diverse pool of sequences for functional lux operons from the environmental species also allows for a powerful directed evolution approach that can robustly explore large part of the sequence space to evolve optimized lux reporters. To this end, we have successfully implemented the DNA-shuffling method and in the process of evolving the decanal-specific luxAB genes, and autonomous lux reporters through the evolution of the restructured lux operons.
For the follow on outreach to the grant, we are collaborating with artists and fashion designers from the RCA to create an art installation to exhibit at the Cambridge e-Luminate festival. In this installation, we will display some of the brightest environmental bioluminescent bacteria in a setting that will allow the general public to engage with our work and create a discussion about the use of biology as technology.
We have submitted the sequencing and assembly data produced as part of of our project to the NCBI public repository and will be available following curation by the NCBI staff. Links are provided on the end of this document. Upon the completion of our evolution experiments, we will publish our quantification data and constructs publicly.